Campi chirurgici
Chirurgia della Base Cranica
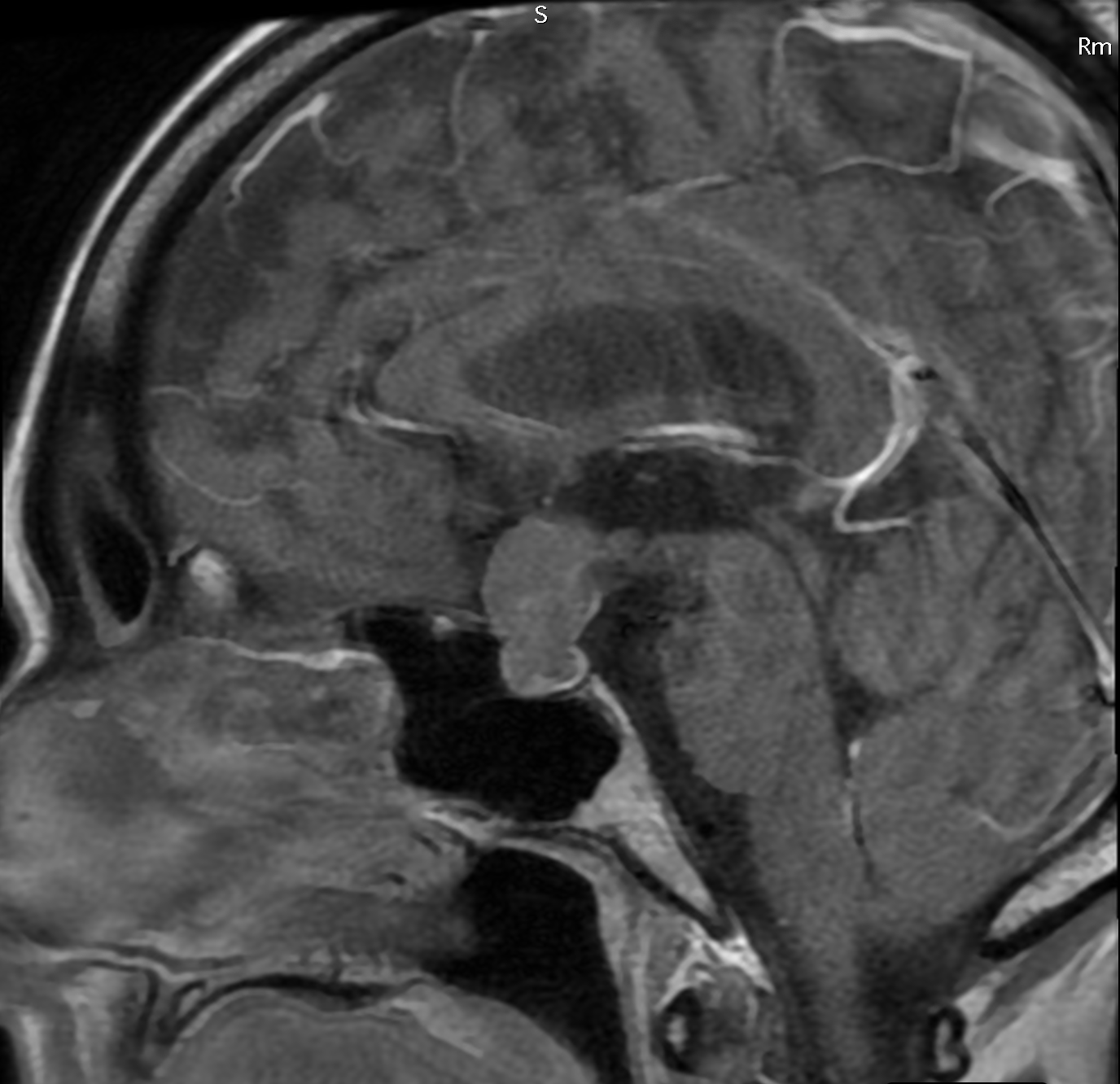
Craniofaringioma
I craniofaringiomi sono tumori congeniti di natura benigna, a lenta crescita, ad origine dai residui epiteliali del dotto craniofaringeo o della tasca di Rathke; talvolta anche dallametaplasia dei residui delle cellule epiteliali squamose di parti dello stomodeo (contribuiscealla formazione della mucosa orale).
Si localizzano a livello della regione sellare e parasellare del sistema nervoso centrale, in stretti rapporti con il chiasma ottico.
Si manifestano con due picchi, uno in età pediatrica tra i 6 e i 16 anni, l’altro in età adulta trai 50 e i 70 anni.
Cilinica e iter diagnostico
I pazienti riferiscono cefalea e disturbi visivi derivanti rispettivamente dalla ipertensioneendocranica e dalla compressione del chiasma ottico che la neoplasia esercita. In base alla crescita del tumore possono manifestarsi sintomi legati alla compromissione di altri nervi
cranici, quali nervi oculomotori con comparsa di diplopia (visione doppia), nevralgie trigeminali e disturbi olfattivi. Altri disturbi riguardano la sfera endocrinologica, quali ritardo nella crescita o ritardo della pubertà.
La diagnosi viene posta mediante esami strumentali quali RM encefalo.
Cilinica e iter diagnostico
I pazienti riferiscono cefalea e disturbi visivi derivanti rispettivamente dalla ipertensioneendocranica e dalla compressione del chiasma ottico che la neoplasia esercita. In base alla crescita del tumore possono manifestarsi sintomi legati alla compromissione di altri nervi
cranici, quali nervi oculomotori con comparsa di diplopia (visione doppia), nevralgie trigeminali e disturbi olfattivi. Altri disturbi riguardano la sfera endocrinologica, quali ritardo nella crescita o ritardo della pubertà.
La diagnosi viene posta mediante esami strumentali quali RM encefalo.
Trattamento
Il trattamento è per lo più chirurgico con finalità radicale, quando possibile. L’approccio sfruttato è soprattutto quello endoscopico transnasale transfenoidale, transtuberculum sovrasellare che permette il raggiungimento della neoplasia sfruttando la cavità nasale, come corridoio chirurgico, rappresentando dunque un approccio mininvasivo.
La radicalità chirurgica può essere ottenuta se non vi è interessamento dell’ipotalamo: in caso contrario è necessario una resezione subtotale seguita da radioterapia post-operatoria.
Considerando la vicinanza con la ghiandola ipofisi, una potenziale complicanza è rappresentata da disturbi endocrinologici che nel postoperatorio possono necessitare di terapia sostitutiva.
Per maggiori informazioni, contattaci
LE ATTIVITA’ NEL DETTAGLIO
Altri interventi chirurgici e patologie trattate
Trattamenti endoscopici delle stenosi tracheali
Questi trattamenti sono utilizzati come prima scelta a seguito della diagnosi di stenosi sottoglottica-tracheale.
Indicazioni:
– Stenosi di basso grado
– Paziente non fit per chirurgia open
– Trattamento “ponte” in attesa di programmazione chirurgica
Scialoadenectomie (rimozione di ghiandole salivari maggiori)
Con il termine scialoadenectomia si intende la rimozione di una ghiandola salivare maggiore, solitamente ghiandola parotide (parotidectomia) o ghiandola sottomandibolare; più raramente può riguardare la ghiandola sottolinguale.
Svuotamento laterocervicale
Lo svuotamento laterocervicale è una procedura chirurgica che prevede la rimozione di alcune vie linfonodali delle stazioni laterocervicali. Secondo Robbins si possono distinguere 7 livelli laterocervicali per lato (solamente il settimo livello è impari e mediano) delimitati da specifiche strutture anatomiche che vengono utilizzate come punti di repere durante l’atto chirurgico.
Lembi peduncolari (lembo pettorale, lembo sovraclaveare,indiano..)
Alcuni interventi di oncologia maggiore e minore sono accompagnati da un tempo ricostruttivo che si basa sull’utilizzo di lembo peduncolati o liberi. I lembi sono tessuti comprendenti muscolo, fasce muscolari o cute e anche osso, tutti irrorati da uno specifico asse vascolare.
I lembi peduncolati sono quelli che mantengono un apporto vascolare dal sito di prelievo e che vengono ribaltati nelle regioni circostanti, mentre i lembi liberi sono quelli che vengono trasposti e poi innestati in un sito ricevente dopo aver eseguito un’anastomosi (unione) dei vasi sanguigni.